- Panel Discussion with all Instructors
- Real World Evidence
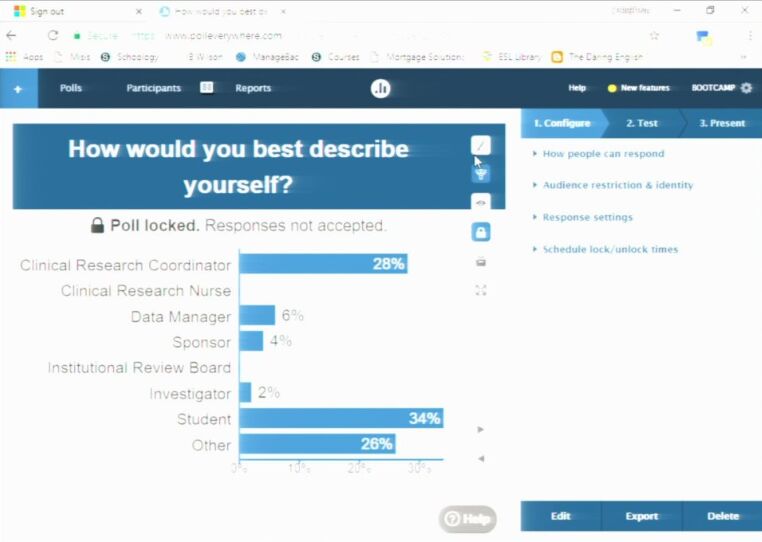
- What are we looking at from a design stand-point?
- Real World Evidence – Idea to put an emphasis on how drugs and products are used in the real world to make the clinical trial process more convenient and condensed
- Generalized variability – benefit of real world evidence
- Post-market surveillance – expected in both the US and Europe; always been an emphasis on this; legacy data may not be sufficient and real-world evidence may be combined with legacy data to produce a sufficient of quality data
- Too-controlled data may not be reflecting real-world effects or cannot be effectively translated into the real world
- The implications of design and regulatory considerations with this growing consideration/use of real world evidence
- Using real world evidence in single arm trial can supply second best control group
- Idea of substituting real world data for clinical trials for certain conditions like drugs with severe ailments
- i. Allowing patients to use these drugs and expand the brand based observations and effects on the first patients using these medications without conducting clinical trials, which can be concerning
- There is a guidance being described for using real world evidence
- Concern: What is the quality of real-world evidence?
- Others argue that clinical trials already reflect real-world because these patients and the effects of the drugs cannot be controlled
- Adaptive Trial Design
- Industry is ahead of regulation
- Are there any concerns with adaptive trial design and other health authorities/regulatory agencies may not be open to it?
- Separate analysis plans per country (recognizing that there are differences with differing countries)
- Opinions of regulatory agencies differ more for devices than drugs
- Sample Size of Clinical Trial/Control Group
- How to Determine the Sample Size of the Control Group
- Equal allocation for both arms in two-arm trial; easier to enroll patients if they know there is 50-50 chance of them receiving the medication; but most of the time, 1:1 is what are designed
- Gold Standard for Devices
- What is the delta? Is this delta significant? The gold standard is comparing your device against another device
- Opinion of Audits, Staff Increase, Suppliers
- Short-term thinking from politicians due to scandals; they are more concerned with saving their site than making sure drugs that have been used for 30 years to remain accessible to patients when these drugs may not meet the new regulations
- Supply chain: New actors in the downstream process, everybody is checking everybody, massive change (Who is auditing them?), people do not want to do it, takes more financial resources to complete the audit
Regulatory Science Symposium: Regulatory Aspects of Clinical Trial Design Session 6: Panel Discussion (2018)
In this session, audience members ask the panel questions concerning real world evidence, the gold standard for devices, adaptive trial design, sample size of control group, and legislation changes in European drug regulations.
Regulatory Science
Clinical Trials

Eunjoo Pacifici, PharmD, PhD
Chair and Associate Professor of Regulatory and Quality Sciences Associate Director, D. K. Kim International Center for Regulatory Science

Nancy Pire-Smerkanich, DRSc, MS
Assistant Professor, USC Mann Dept. of Regulatory and Quality Sciences; Associate Director, Regulatory Knowledge and Support

Steven Snapinn, PhD
Amgen

Steven Wilson, DrPH
Center for Drug Evaluation and Research-Retired 2017
Course Syllabus/Topics
Acknowledgement
Accompanying text created by Annie Ly | Undergraduate Research Associate and Provost's First Generation Research Fellow | lyannie@usc.edu